 "
"
Team:GenetiX Tec CCM
From 2014hs.igem.org
Welcome to our wiki
We are high school students at Tecnológico de Monterrey, Mexico City Campus, a team with juniors and seniors working together. What made us a team was our passion for science, we all wanted to innovate, to create, to surprise and engine something useful. Our goal is to prove that if you plan on doing something, no matter what, you can achieve it with the right focus.
Biodetection of Anoxia in Lake Xochimilco
Lake Xochimilco in Mexico City faces a condition of extreme pollution which endangers the endemic species; many of which are nearing extinction. Oxygen levels depletion in the lake directly affect the fauna, making it less hospitable or even deadly. Our goal is to produce a biosensor that can easily and inexpensively detect anoxia in different regions of the lake. Using an oxygen promoter in addition with the biological markers RFP and GFP we could theoretically detect low dissolved oxygen levels in water samples. In addition, we intend to use a second construct with an Iron promoter to detect iron concentrations that also endanger the sustainability of living organisms in the lake. Once we identify critical regions of the Lake, our report could incentivize the Civil Council and authorities to propose concrete legal initiatives to reduce pollution in the identified areas and start remediation campaigns.
Contents
The Project

The main idea of our project is to achieve the detection of anoxia and Iron concentrations in water systems. What we propose is to construct an easy way of monitoring the levels of O2 and Fe in the lake by using biosensors. By using modified bacteria E. coli for this, we will try to find a cheaper, easier, and faster way to detect the problem of anoxia and iron concentration in some aquifers of Mexico City. We will have to analyze samples of water at different depths to know where the problem is worse and what probable native species could be more affected.
Our purpose is the identification of adequate dissolved oxygen
levels for a stable support of life and the identification of iron
concentration below threatening levels in order to know if the
lake has the physical and chemical properties to support wild-life
naturally. With the use of biosensors, specialized for detecting
concentrations of oxygen above a 2% dissolution, we used modified
E. coli with an oxygen promoter (BBa_K258005) that will
detect the low concentrations and a reporter of (GFP or RFP) that
will indicate the activation of our promoter (Figure 1).
The Iron promoter reacts inside an environment with a
concentration of iron ranging from 1 ppm and on (A.
Quintero,2007). The acceptable levels of iron in drinkable water
are lower than 0.5 ppm (WHO, 1996).

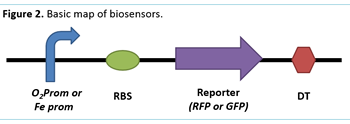
To achieve the objective using E. Coli we will construct different types of modified plasmids for our bacteria to express the biosensors based on iGem biobricks. The idea is to use sensitive promoters: one for oxygen, and another one for iron; those promoters will lead to an expression of GFP or mRFP. This will provide a visual signal to indicate the presence or absence of these elements. Biobrick parts BBa_K258005 (O₂ prom), BBa_I765000 (Fe prom), BBa_E1010 (GFP) and BBa_J04650 (mRFP) were selected for the construction of the biosensors (Figure 2). These were transformed into E. coli strands DH5-a, TOP 10, and NEB 10-b for storage and subsequent plasmid growth and isolation using a Zymo Research® DNA extraction kit.
Once we measure and identify critical regions of the Lake, our report could go directly to the Citizen Council for its consideration. The competent authorities should be able to propose concrete legal initiatives to reduce pollution in the identified areas and start remediation campaigns that re-establish the local aquatic environment to a stable, liveable, friendly ecosystem for the inhabitant species.
Biosensor

A biosensor is an instrument for the measurement of biological or chemical parameters. They usually combine biological and physical-chemical components.
Generally, they consist of three parts:
- The biological sensor: It may be a tissue, a culture of microorganisms, enzymes, antibodies, nucleic acid chains, etc. The sensor can be taken from the wild or be a product of synthetic biology.
- The transducer: Its function is to bind the other two elements and translate the signal emitted by the sensor.
- The detector: It can be optical, piezoelectric, thermal, magnetic, etc.
The most common example is a biosensor that measures blood glucose. It uses an enzyme that processes glucose molecules, releasing an electron for each molecule. Said electron is collected at one electrode and the electron flow is used as a measure of the glucose concentration.

The caged canaries used by miners to detect the presence of lethal
gases can be seen as an early example of biosensors (Wikipedia,
2014)
By using modified bacteria E. coli for this, we will try to find a
cheaper, easier, and faster way to detect the problem of anoxia
and heavy metals in some aquifers of Mexico City. We will have to
analyze samples of water at different depths to know where the
problem is worse and what probable native species could be more
affected.
Some of the benefits of using biosensors instead of other sensing
methods, as observed by Ajit Sadana and AzoSensors, are:
- A fast response in time.
- Fast and continuous measurement.
- High specificity because of its shape-specific recognition.
- Simplicity in its use.
- Capability of measuring concentrations ranging from 10-18 to 10-19 M, so we need low sample requirements.
- Capability of real time measurements.
Eutrophication

Eutrophication is the process by which the increased availability
of one or more limiting growth factors needed for photosynthesis
cause excessive plant and algal growth. Some of these factors are
the amount of carbon dioxide, sunlight and nutrient fertilizers.
The elements coming from the nutrient fertilizers that especially
affect the photosynthesis rate are nitrogen and phosphorus.
(Chislock , 2013)
Plants require many different nutrients or components for the
realization of photosynthesis. Nitrogen and phosphorus are the
first components depleted in the water even though there is a
greater amount of other needed substances. While performing
photosynthesis about 8 times more nitrogen is needed than
phosphorus. Thus, phosphorus limits eutrophication if nitrogen is
more than 8 times abundant as phosphorus, while nitrogen is the
limiting factor when its concentration is less than 8 times
abundant as phosphorus. Erosion of surrounding areas is also an
important cause of eutrophication because the nutrients of the
ground are not retained by the roots of plants and trees that
should be there. So deforestation is an environmental element that
strongly affects this process. (UNEP, 1)
The process of eutrophication of an aquifer occurs naturally over
centuries as they are filled with sediments, abundant in nutrients
(figure 1). However, this process has been recently much
accelerated due to the contamination produced by human activities.
The discharges into aquatic systems bring a lot of limiting
nutrients for eutrophication, including nitrogen and phosphorus.
These polluting human residues thrown up into water systems come
from point and non-point pollution sources. (Chislock, 2013)

Figure 1. Natural eutrophication
The term “point source” is referred to
as any single, discernible source from where the polluting agent
is originated, such as a discharge pipe from a factory, sewage
plant. The other term “non-point source” means that the pollution
does not come from a single determinate source. This type of
pollution happens when water moves across the land and pick in its
way human-made pollutants that can be deposited later on in water
bodies. (Harvey, 1)
There are different levels of eutrophication according to how
severe or advanced the process is. The first and harmless
classification of eutrophication is the oligotrophic, where there
is a low concentration of nutrients in the water and thus less
biologic production. Then we have the mesotrophic where there are
intermediate levels of nutrient concentrations and there is a
moderate biologic production that doesn´t affect severely the
aquatic environment. The real problem begins when we get to the
eutrophic level where there is an elevated concentration of
nutrients and a very high biologic productivity. Another
classification is reserved for where the nutrient levels reach
extremely dangerous concentrations that take the aquifer´s
condition to a critical state; it is called hypertrophic and is
almost always caused by the cultural eutrophication. An important
indicator for the eutrophication level is chlorophyll. The total
amount of chlorophyll represents about 1% of plant biomass, so in
this way the total biomass can be estimated allowing the
determination of the degree of eutrophication. (Mazzeo, 1)

Table expressing the characteristic values for each of the
eutrophication classifications. (UNEP)
Eutrophication brings a lot of complications to aquifers. The
enormous creation of dense blooms of noxious, foul-smelling
phytoplankton reduces water clarity and harms water clarity. These
blooms limit light penetration to the water body. This limiting of
sunlight to littoral zones causes the die-offs of the great amount
of plants and algae that grew up without control due to
eutrophication. When these dense algal blooms eventually die,
microorganisms start the decomposition of organic matter and
severely deplete the available dissolved oxygen, causing hypoxia
or even anoxia. These hypoxic environments are cause of dead zones
for most of the inhabiting organisms for the lacking oxygen.
(Chislock 2013)
The normal levels of dissolved oxygen in water for the maintenance
of life are around 6mg/L. Environments are considered hypoxic when
the concentration of dissolved oxygen goes below 2.8 mg/L. When
the dissolved oxygen levels reach the hypoxic condition many
species die. Depending on the size and other characteristics of
the organisms, the limiting concentration for survival will have
low variations. The hypoxic conditions can change in different
lapses of time. They can occur just for a few moments
(minutes/hours) or they can reach chronic states that last for
weeks or even months, causing depletion of local species.
(Cisterna 2008)
It is important to supervise aquatic environments conditions´ to
prevent the initiation of eutrophic conditions. Eutrophication can
kill all life in natural environments. If some symptoms of
eutrophication are detected in time it is possible to attack the
problem and control it, or even eliminate it. Some methods for
controlling eutrophication are:
- Covering sediments, preventing release of nutrients.
- Biomanipulation
- Using chemicals such as copper sulfate to kill excess of algae
- Aerating the hypolimnion of a lake, reducing the release of nutrients from the sediments.
(UNEP)
Localization

First of all, we have to know what eutrophication is, well the eutrophication is the process of excessive growth of algae and weeds water in the water, caused by phosphates and other pollutants discharged to waters. In a eutrophic aquatic ecosystem two things happen: more oxygen is required to break down and increases the population of organisms known as primary producers: organisms that make photosynthesis, as macroalgae and lilies. These can reach atrophy processes exchange of oxygen and water flow. The liquid is cloudy and the lack of oxygen can devastate populations of various organisms.
How does it affect the lake of Xochimilco?
It is estimated that close to urban centers or agricultural nutrient input to a lake can be accelerated by the activities human, a process known as cultural eutrophication, which is the case with Lake Xochimilco to be within the city of Mexico. This is precisely effluent mainly caused by plants sewage treatment, containing nitrates and phosphates runoff of fertilizers and waste animals and accelerated erosion of eutrophication .The eutrophication derived from crops by the recent addition of phosphates and nitrates, as a result of activities human, it is also a serious problem for lakes, especially Lake Xochimilco. During warm periods, the overhead produce dense growth nutrient vegetables such as algae, cyanobacteria, water lilies and duckweed. Oxygen dissolved in the surface layer of water is near die exhausted when large masses of algae, which fall to the bottom and are decomposed by aerobic bacteria. This can kill fish and other animals water they consume oxygen. If theexcess nutrients continue to flow a body of water, the water reaches the bottom be rotten and almost unusable for living things, because bacteria take over and produce anaerobic decomposing substances with poor odors, such as hydrogen sulfide and the methane.
Factors
The factors affecting the degree of eutrophication are:
- Climate: warm climates favor the process.
- Shallow bodies of water and / or low flow are more conducive to the development process
- Drainage Area: little tree cover subject to abundant rainfall tions favors erosion and entrainment of nutrients into the water body
- Geology: In drainage areas where sedimentary rocks predominate vapor no greater phosphorus runoff. Clay soils drain poorly and also favor runoff and result in nutrient supply.
The causes of eutrophication include:
a) Natural:
- atmospheric inputs: precipitation.
- resuspension of bottom sediments.
- release from anoxic sediments.
- breakdown and excretion of organisms.
- nitrogen-fixing microorganisms.
b) Anthropogenic:
- Discharges of industrial, agricultural, municipal and waste treatment plants.
- Deforestation increases erosion and reduces the recycling of nutrients in the watershed, increasing their income to the water body.
- Fertilizer applied in excess.
- Sewage farms (silos, drums).
- Septic tanks.
- Detergents with large amounts of phosphorus.
- Contribution of pollutants from rainwater.
- Sewer system of cities and towns do.
- Measures to control eutrophication include:
Control of nutrient inputs:
- Waste treatment before being poured into the body of water.
- Restricting the use of phosphate detergents.
- Control of land use.
- Prepantanos: remove nutrients from waste water that are fixed in the biomass of algae and macrophytes.
- Physical and chemical waste water treatment: chemical precipitation and filtration.
What we found....
Elements found in the lake of Xochimilco
| Sample | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| MO mg/LO | 154 | 251 | 88 | 151 | 555 | 295 | 25 | 75 |
| Phosphorus mg/L | 5.8 | 5.6 | 7.6 | 6.7 | 23.0 | 17.1 | 15.0 | 14.2 |
| pH | 6.2 | 7.2 | 6.7 | 7.6 | 8.0 | 7.0 | 7.0 | 7.0 |
| Temperature °C | 3.8 | 3.8 | 4.8 | 3.8 | 6.9 | 7.0 | 7.0 | 7.0 |
| Precipitation | 21.0 | 20.0 | 22.0 | 23.0 | 21.5 | 19.8 | 21.0 | 54 |
Results

Construction of the proposed biosensors was not completed successfully due to what appear to be flawed ligations and/or low efficiency competent cells. The necessary methodology and procedures have been laid down for further work on construction, development and improvement of the biosensor system in the future. Further work include the completion of the biosensors, proof of concept growing bacteria in absence and presence of stimuli and application of biosensors to a range of samples from specific points distributed all along Xochimilco lake. Results of these tests could be then applied in the construction a map of the lake specifying the most critical points that need to be treated.

Figure 1. The agarose gel shows the unsuccesful ligations, represented on the right.
Human Practices
Travesuras 2014

GenetiX, as a student group, participated in the event Travesuras 2014, which had place the 26 of April 2014 inside the installations of Tecnologico de Monterrey, Campus Ciudad de Mexico. The GenetiX station was located in the high-school chemistry laboratory where 4 groups attended, composed around of 25 children each, with ages that ranged between 7 and 16 years. Inside the station there were nine of our members present involving children into science in a fun and enriching way for them. These experiments included de polymerization of “Slime” with color and “Silly Putty”.

Before initiating the experiment, a quick explanation of the
process of polymerization was given. For the polymerization of the
Slime, the kids had the opportunity of adding in plastic cups 10mL
of polyvinyl alcohol and 3 mL of a borax solution in addition with
a drop of colorant. Later, the children agitated the mixture with
wooden sticks until it took the slimy consistence expected. This
procedure was made in groups of between 2 and 3 kids who shared
the slime.

A polymerization of the silly putty was made in the same way in which the slime was made. In this case, 10 mL of white glue (which contains the polyvinyl acetate) and 20 mL of saturated borax solution. The children agitated the mixture until it obtained the desired consistency. Later, they were able to play with it also in groups of 2 or 3 children. All the experiments were supervised by the members of GenetiX in order to prevent any accident and solve further doubts from the children regarding the experiment.

Finally a DNA demonstration took place. For such, a banana was cut
into pieces and got liquefied in a blender with distilled water
and 2.5 gr of NaCl. It was liquefied until a dense liquid was
achieved and then poured into a coffee filter inside a glass
flask. We left the mixture in filtration for about 10 min until
the flask was half-full. Afterwards 5ml of liquid soap was added
to the pulp in order to break the cell walls and membranes and
free the DNA. Also 3 gr of meat tenderizer and another 3 grs of
sodium bicarbonate were added into the mixture. Once the
ingredients were added, the sufficient amount of cold ethanol was
added in order to form a thin layer above the banana mixture.
Since the DNA is insoluble in ethanol, a whitish layer appears
above the banana mixture. Once the process ended, the final
mixture was shown to the children and a brief explanation of what
DNA is was given.

Travesuras 2013

GenetiX firstly participated in the Christmas themed Travesuras 2013. In this event, kids from other schools raging from Elementary schools to Middle Schools attended to be part of various activities made by us, students from the Tecnologico de Monterrey, surrounding the topic of Christmas.

We were mostly involved in helping prepare the decoration of the main stations. Some of the activities that we participated in was the making of cardboard spheres and decoration of said materials, including decorations such as Christmas trees, hanging colored paper in patterns and the working tables as well.

We also helped to cover the floor for decoration and prevent further stains in the real floor. We were tasked with watching and guarding the kids during the whole event and also to entertain them.
Conference

As part of our Human Practices, on June 14th 2014, we gave a presentation for students and teachers from Tecnológico de Monterrey, Colegio Lestonnac and other Middle School and High School level institutes. As students and teachers who are interested in Science were invited, their participation was very dynamic. There, we explained basic concepts of Synthetic Biology and Biotechnology to the audience.

Our main topic was Synthetic Biology and its present and future
applications. But, in order to transmit our message clearly, we
had to start from the basics; that is why, one of our contents
(the first one to be presented), were the characteristics of
living beings. In this point, we stated that a living being has
complex and organized systems, have a metabolism, need to maintain
homeostasis, can grow, can reproduce, respond to stimuli, and are
adaptable.

We gave the definition of Synthetic Biology as the design and
construction of biological parts, devices or systems, or the
re-designing of existent natural biological systems in order for
them to be useful, and we also explained the techniques for the
creation of synthetic organisms, such as acceleration of heavy
particles, electroporation or heat shock. In order to
complement the concepts, we presented some examples of synthetic
organisms and their functions, such as water pollution-absorbing
plants, trees that change their color in case of a chemical attack
and goats that can produce spider web.

Afterwards, we explained our participation in the iGEM contest,
described our project to the audience (who found it interesting
and useful) and performed an experiment which consisted in
extracting DNA from bananas to stimulate their scientific
curiosity.

Data
Safety
Important Definition:
Biohazard: An agent of biological origin that has the capacity to produce deleterious effects on humans, i.e. microorganisms, toxins and allergens derived from those organisms; and allergens and toxins derived from higher plants and animals.
Biosafety: The containment principles, technologies and practices that are implemented to prevent the unintentional exposure to pathogens and toxins, or their accidental release
Biosecurity: Control of accidental and deliberate release of biohazardous material
Safety Questions.
- Would any of your project ideas raise safety issues in terms of
- Researcher Safety: All team members had to participate in a general health and safety induction, where we learned handling biological material, aspects on chemicals, guidance in waste disposal of sharps, trace chemicals, and biohazardous material and the general protocols of the lab we work in. At all times while working in the lab, we were supervised by our advisor, part of the High school Science Department, or lab technician from the university’s School of Life Sciences Laboratory.
The lab we work in is classified as BSL 1 (biosafety level 1), according to the European Union Directive 2000/54/EG. Work inside a BSL 1 lab does not involve the use of potentially harmful materials to the researchers if they act corresponding to the general precautionary measures. Researcher should wear a lab coat, safety glasses and gloves and one must not drink, eat or smoke whilst working at the bench. The safety degree of the worn protection should depend on the chemicals and microorganisms handled. An important example is handling antibiotics and DNA coloring agents with gloves and safety googles. Most importantly, everybody should always be aware of what he is doing, with what kind of biological parts and chemicals he is working and how to handle them safely. - Public Safety: We used different bacterial strains throughout our project. E. coli 10 beta, Top 10 and DH5 alpha; which are non-toxicogenic, disabled, non-pathogenic, non-colonising, laboratory-adapted K12 strains, which are widely used in research and present no hazard to human health.
- Researcher Safety: All team members had to participate in a general health and safety induction, where we learned handling biological material, aspects on chemicals, guidance in waste disposal of sharps, trace chemicals, and biohazardous material and the general protocols of the lab we work in. At all times while working in the lab, we were supervised by our advisor, part of the High school Science Department, or lab technician from the university’s School of Life Sciences Laboratory.
- Environmental Safety:
In the lab, waste must be contained in a biohazard box with an autoclavable biohazard bag. Liquids must be inactivated either via chemical methods (e.g., with bleach) or using an autoclave. Solids that have been in contact with biohazardous materials should also be treated by autoclaving and then transfered into a different bag to indicate that the waste has been deactivated. Broken glass and needles should be disposed in a sharps container. Full and sealed sharps containers can be added to solid waste.
References:
Chart, et al (2000). An investigation into the pathogenic properties of Escherichia coli strains BLR, BL21, DH5a and EQ1. Journal of Applied Microbiology, 89, 1048-1058. URL: http://ors.uchc.edu/bio/resources/pdf/3.6.1.A_colipath.pdf
Escherichia coli K-12 Derivatives Final Risk Assessment. Biotechnology Program under the Toxic Substances Control Act (TSCA). URL: http://epa.gov/biotech_rule/pubs/fra/fra004.htm
- Do any of the new BioBrick parts (or devices) that you made this year raise safety issues?
- Did you document these issues in the Registry?
- How did you manage to handle the safety issue?
- How could other teams learn from your experience?
No extra safety issues were detected during the construction of our biosensor. The secondary ecological consequences will have to be assessed once the system is applied.
- Is there a local biosafety group, committee, or review board at your institution?
What does your local biosafety group think about your project?
The school has a person in charge of Health and Safety for each of the Laboratories. The technician was in constant contact with the team, supervising that the rules were being followed.
We also adhered to our countries legislation in the Chamber of Deputies (the Law of Biosecurity for Genetically Modified Organisms, Nueva Ley DOF-18-03-2005 http://www.diputados.gob.mx/LeyesBiblio/pdf/LBOGM.pdf), the SAGARPA and the Law of Science and Technology from CONACyT.
- Do you have any other ideas how to deal with safety issues that could be useful for future iGEM competitions? How could parts, devices and systems be made even safer through biosafety engineering?
In the assembly and distribution kit the iGem could include safety equipment such as a Laboratory Health and Safety Manual, gloves and googles samples. In the case of our project, we could include two resistance markers so the bacteria, if accidentally released, had less probability of subsisting.
Notebook
Protocol 1
Preparation of culture media (1L). 1.- 5 g of yeast 2.- 10 g of sodium chloride. 3.- 10 g of triptone. 4.- Add water till 1 uL volume.
Protocol 2
Competent cell´s preparation 1.- Centrifug your media culture tube (25 mL) at 4,000 rpm during 15 min. 2.- Pour the supernatant and add 10 mL of cold MgCl2 solution. Centrifuge them at 4,000 rpm at 4 °C during 15 min. 3.- Pour the supernatant and resuspend the pellet in 40 mL of cold CaCl2 solution. Keep it in ice during 20 min and place them in eppendorf tubes. Centrifuge them at 4,000 rpm at 4°C during 15 min. 4.- Pour the supernatant and resuspend the pellet in 40 mL of cold 85mM CaCl2/15% glycerlol v/v solution. Centrifuge again at 2,100 rpm at 4°C during 15 min. 5.- Pour the supernatant and resuspend in 2 mL of cold 85bmM CaCl2/15% glycerol v/v solution. 6.- Aliquote in tubes of 100 uL previously frozen and store at -80°C.
Protocol 3
DNA extraction from Buffy coat: 1.- Centrifuge 5 mL of culture medium at 1,500 rpm for 10 min at 4°C. 2.- Carefully transfer 600 uL of buffy coat to a microtube. 3.- Add 1 mL of RBC buffer and mix throughly by vortex. 4.- Centrifuge at 4,500 rpm during 30 s and pour carefully so your pellet is not wasted. 5.- Repeat steps 2 and 3 adding just 500 uL from RBC buffer. 6.- Once separated, add 300 uL of lysis buffer to the pellet and mix by vortex. 7.- Add 150 uL of protein precipitation solution. 8.- Centrifuge at 11,000 rpm during 5 min; then, transfer the supernatant to a new microtube. 9.- Add 700 uL of cold isopropanol and mix by inverting until you see a white formation within the tube. 10.- Centrifuge at 11,000 rpm during 4 min and pour carefully taking care of the pellet at the bottom. 11.- Add 500 uL of cold ethanol 70% to wash and then centrifuge at 11,000 rpm during 4 min. 12.- Pour and let the ethanol evaporate at room temperature. 13.- Resuspend the DNA pellet with 12 uL of TE buffer; incubate all night at 4 °C.
Protocol 4
Electrophoresis: 1.- Prepare 1L of TAE 10X (48.4g of Tris base[ tris (hidroxymethyl) amminomethane], 11.4 mL of glacial acetic acid (17.4 M) and 3.7 g of EDTA then fill with deionized water to 1 L. 2.- Prepare a 50 mL solution of 3% agarose using TAE buffer (without using water). 3.- Heat the gel in the microwave in intervals of 30 s, 20 s and 10 s until the agarose is completely dissolved, taking care that it does not boil so much. 4.- Let cool down the gel (55-60°C) and pour in a base with the comb already on its place to form the charging wells. 5.- Wait until the gel solidifies. 6.- Remove the comb and place the base with the gel inside the electrophoretic chamber. 7.- Add TAE buffer until the gel is covered. 8.- Take 5 uL of each sample from the PCR and 1 uL of charging gel in a new PCR tube, mix well. 9.- Charge the samples inside the wells of the gel, adding a weight marcker. 10.- Close the electrophoretic chamber and run at 120 V during 30 min. Follow the displacement ofthe samples. 11.- Take out the gel from the chamber very carefully and place in the UV light to see the DNA strands.
Protocol 5
Competent cell transformation: 1.- Place 1 uL of the plasmid in a microtube, gently add 100 uL of your competent cells. Take another tube and place only 100 uL of competent cells, so you have a control group. 2.- Mix gently with a pipette and incubate during 30 min in ice. 3.- Ab ruptly place your cells in 42°C dry bath during 2 min and replace in ice whwn finished. 4.- Add 900 uL of LB medium and incubate at 37 °C for 30 min. 5.- Inoculate with 200 uL of your transformed cells in LB-Amp. 6.- Incubate overnight and store at 4 °C of freeze in 15-25% glycerol.
Lab archives// Mar 12, 2014
In order to grow our bacteria, we inoculated with 100 uL of E. Coli DH5-alpha in 50 mL of LB medium, and 100 uL of E. Coli TOP in 50 mL of LB medium. We left both samples incubating overnight
Lab archives// Mar 20, 2014
We inoculated E.Coli NEB10-beta in 50 ml of LB medium.
Lab archives// Mar 27, 2014
We prepared the medium in which our bacteria will grow, for that we dissolved solid LB with agarose, then we add it into 10 plates, we waited a few minutes for it to solidify and finally we added 500 uL of our antibiotic that is chloranphenicol to each of those plates.
Lab archives// Apr 01, 2014
we transformed luciferaze from DNA to cells by incerting them with change of temperature
Lab archives// Apr 02, 2014
We made a litter of agar and 500 L of growth medium, and we sterilized them along with crystal media disperssion balls
Lab archives// Apr 03, 2014
Today we prepared buffers for home made mini preps and did a cell re culture.
Lab archives// Apr 07, 2014
We added 700 mL of water in a beaker and then we prepared 23 grams of powdered agar that we added slowly while we moved the beaker. This in order to help the dissolve the powder. After that, we added other 300 mL to the solution and finally we passed it to an Erlenmeyer matrass. Also we prepared 500 mL of growth medium. First, we measured and added 5 grams of tryptone, 5 grams of yeast and 10 grams of sodium chloride. Those ingredients were added slowly to a graduated cylinder with 350 mL of wáter while the flask was moved. After finishing the addition we added other 150 mL of water. The solution was then transfered to another Erlenmeyer flask. Both Erlenmeyer flasks and a glass bottle with crystal media diperssion balls were sterilized with temperature and pressure.
Lab archives// Apr 02, 2014
300 ul of ampicillin at 100mg/ml chloramphenicol 3 Plates with agar Promoters: BBa_K258005 (pO2), BBa_176500 (pFe) BBa_J52008 (luc-3) The plates were impregnated with ampicillin promoting bacteria in the following manner |pO2 | Plate | 150 ul Ampiciline | K258005 | ------------------------------------------------------ |pFe | Plate | 150 ul Ampiciline | 1765000 | ------------------------------------------------------ |Luc-3 | Plate | Chloranphenicol | J52008 |
Apr 08, 2014
**/Today we realized the mini prep for the oxygen promoter. /** --500 ul of Ampiciline //this is for the oxygen promoter --50 ul of oxygen promoter --45 ml of LB medium --at room temperature 15 minutes at 1500 rpm in the centrifuge. --500 ul of chloramphenicol // this is for the red liciferase --50 ul of red luciferase --45 ml of LB medium --The new LB medium was prepared with //LB medium --10 g yeast --5 g salt --15 g Tryptone MINI PREP First 400 ul of Genomic Lysis Buffer to the 40 ml overnight [bacteriaand oxygen promoter] Then the mixture is transferred to a Zyne-Spine Collumn in a Collection Tube. Centrifuge 10,000 rpm /1 min Transfer the Zyno Spin Column to a new Collection Tube. Add 200 micro liters of Pre Wash Buffer. Centrifuge 10,000 rpm /1 min Add 500 uL of of g-DNA Wash buffer to the spin column. Centrifuge 10,000 rpm /1 min Transfer the spin column to a clear micro centrifuge tube. Add 50 uL of DNA Elution Buffer . Incubate 2-5 minutes to at room temperature. Centrifuge at top speed per 30 seconds. Then store at -80 °C
Apr 22, 2014
We prepared 2 overnight cultures (45ml of LB medium): GFP E0040 pSB1A2 with 100 uL of ampicillin. M-cherry (RFP) BBa_J04450 pSB1C3 with 500 uL of chloramphenicol.
Apr 24, 2014
2 Transformations: Bacteria NEB 10 Beta, DH5alpha 2 Mini Preps: GFP, RFP //Transformation and preps:-- Centrifugation at 2500 rpm for 15 min.--1ul DNA in 100 uL of bacterial culture (luciferase). --Filtering GFP, RFP bacteria. --Added 400 ml of Lysis Buffer Genomyc each crop by columns. --Centrifugation at 10,000 * g for 1 min. --Last column to another tube and DNA Prewash + 200 uL Buffer. --Centrifuged at 10,000 * g for 1 min. --Add 500ul of DNA Wash Buffer, centrifuged at 10,000 * g for 1 min. --Transfer to clean tube, add 50ul of DNA Elution Buffer. --Incubate for 2-5 min at room temperature, centrifuged at maximum speed for 30 sec.
Apr 28, 2014
Today was prepared LB media. It also was done the sterilization of 1 mL and 200 uL tips, water and ependorfs. After the sterilization of the LB media was done, we cultivated in it the bacteria carrying the oxygen promoter, RFP, and GFP. Then we left the bacteria growing in the incubator at 37°C overnight.
May 05, 2014
Plating bacteria with GFP, RFP, promoter Oxygen and iron promoter plasmids. Everyone in their respective environments and specific antibiotics (ampicillin and chloramphenicol). Tubes with bacteria was added and placed them in LB medium incubator to grow more product if necessary.
May 08, 2014
We did a centrifuge at 4800 rpm for 20 min for the plasmids had been in overnight which had oxygen promoter, GFP and RFP to rush after we separate the aqueous phase.
May 19, 2014
We did inoculation of GFP, RFP and Oxigen Prmoter
May 20, 2014
We started minipreps preparation of GFP, RFP and Oxigen Promoter. We added 1.5 ml of culture to each Eppendorff (4 for each promoter) and we centrifugated it at 13.2 rpm for 10 minutes, we did the same procedure 2 times. Also, we sterilized two Erlenmeyer flasks and 40 Eppendorffs.
May 20, 2014 //Mini prep preparation
1. We added 1.5 mL of the cultures to their corresponding Eppendorffs and centrifugated them at 3400 rpm during 10 minutes other three times.
2. Once we had a pellet we discarded the supernatant and we added 300 uL of TEG buffer (made of 100 mM of Tris-HCl pH8, 2 mM EDTA and 20% glucose)
3. We added 700 μL of NS solution (0.2 M NaOH, 1% w/v SDs) and we mixed by inversion.
4. We added 400 μL of 3 M sodium acetate pH 5.3, and we mixed by inversion.
5. We centrifugated the Eppendorffs at 3400 rpm during 15 minutes.
6. We transfered the supernatant to other Eppendorffs and we added 50 uL of cold isopropanol and mixed by inversion.
7. We centrifugated the Eppendorffs at 3400 rpm during 15 minutes.
8. We discarded the supernatant and washed each pellet with 70% ethanol and we let it dry for 15 minutes.
9. We resuspended the pellet in 500 uL of TE buffer and then we added 10 mg/mL RNAseA.
11. We precipitated the DNA with 100 uL of ethanol in each Eppendorff.
12. We centrifugated for 1 minute at 3400 rpm.
13. We got rid of the supernatant.
14. Finally, we resuspended the pellet in 50 uL of TE buffer

UNAM procedures// Digestion
|BSA | 1 uL |
|DNA | 2 uL |
|Buffer (2) | 1 uL |
|Buffer Xba I+Pstl | 3.3 uL |
|fe Xba I+Spef | 3.3 uL |
|H20 | 5.4 uL |
Gel agarose
1 kb NEB Ampyciline -> Pentneryl
| ---------------Bristol - Myers Squibb
| ---------------1 gram/ 3 mL
| ---------------inyectable
|pO 2 | 3.3 uL
|1 kb marker 5.4 uL
Homemade Mini prep Preparation
1.- Overnight bacterial culture of 5 mL in falkons of 50 mL
2.- Centrifugate in an eppendor of 1.5 mL and throw supernadant
3.- Add 152 uL of Buffer 1 + RNAse (11 micro-liters /1 buffer micro-liter ) **Buffer 20% Glucouse
4.- Mix via Buffer and add 150 uL of Buffer 2 *Wait 4-5 minutes*
(1.5mL)Buffer 2 -> 880 uL H2O,20uL NaOH 10 N
---------------------------------------100 uL SDS 10%
5.- Mix via inversion and add150 uL Buffer 3
------------------------------Sodium Acetate 3M pH 5
6.-Centrifuge 5 minutes.
7.-Take the supernadant to antoher tube. Add 1 mL ETOH Absolute
8.-Centrifuge by 5 minutes and decant
9.-Wash with 500uL ETOH 70 %
10.-Centrifugate 1 minute
11.-Decant and dry at 37 celsius centigrades (30 minutes)
12.-Resuspend in 15 uL of H2O
Ligation
Inserto 7mL
Plasmid 3mL
Buffer 10x 1.5mL
Ligase 0.5 mL
H2O 3 mL
Total = 15 ml
E.Coli cells transformation.
To transform E.Coli cells with an exogen DNA, 200 uL of competent cells were taken they were mixed with 1-20 ug(Micrograms) of DNA that was incubated on ice for 30 minutes. This makes posible the absorbtion of the exogen DNA to the cells surface, after that, the cells are incubated at 42Celsius Degrees during 90 seconds, for then incubate them during 1 minute again in ice. THis termal shock makes posible the entrance of the plasmid to the inside of the cell. 600 uL of LB medium were added and then incubated during 60-90 minutes at 37 Celisus Degres, to do posible the segregation of the exogenic plasmid during the celular division (3 to 5 generations). Finally, Alicuote of 100-200 uL were cultivated by dimention in the LB agar medium, which ere incubated at 37 Celsius Degrees. After 15-20 hours of incuvation colonies could be observed
Ligations
1# Grow h mL in overnight
2# Do minipreps
3# EcoRI and PstI restrictions.
--for Fe EcoRIand SpeI
O2+ GFP ---1,2,3 - 931
Fe+GTP ---4,5,6 -1,828
O2+ RFP ---7,8,9,10,11,12,13 -890
Fe+RFP ---14,15,16,17,18,19 -1,787
GFP -> 761 bp EcoRI Po+I // 728 bp XbaI SpeI
RFP -> 714 bp Xba - SpeI // 747 bp EcoR - Ps+I
pFe -> 1,067 bp EcoRI - SpeI
pO2 -> 178 bp EcoRi SpeI // 145 bp Xba - Ps+I
O2 + GFP 1/1 - 1/2 - 3/3
FO 2 + GFP 4/1 - 5/2 - 6/3
O2 + RFP 7/1 - 8/2 - 9/3 - 10/4 - 11/5 - 12/6 - 12/13
Fe + RFP 13/1 - 14/2 - 15*16/3 - 17/4 - 18/5 -19/6
Digestion
2 --- BSA --- 1uL
4 --- DNA --- 2uL
3 --- (M) Buffer // Enz (0.3 uL each one)--- 1uL
5 --- EcoRI --- 0.3 uL
--- PstI --- 0.3 uL
1 --- H2O --- 5.4 uL
Total == 10 uL rxn
GFP 761 bp
RFP 742 bp
pFe 1,067 bp
pO2 -15.5 bp
pO2 + GFP 931 bp
pFe + GFP 1,828 bp
pO2+ RFP 890 bp
pFe + RFP 1,787 bp
Gel 1 = 8/9/10/Lodder 1kb/11/12/13
Gel 2 = 15/16/17/Lodder 116/18/19
Gel 3 = 1/2/3/4/Lodder 1kb/5/6/7