 "
"
Team:CSWProteens/notebook
From 2014hs.igem.org


L A B N O T E B O O K:
2013: November | December | 2014: January | February | April | May | June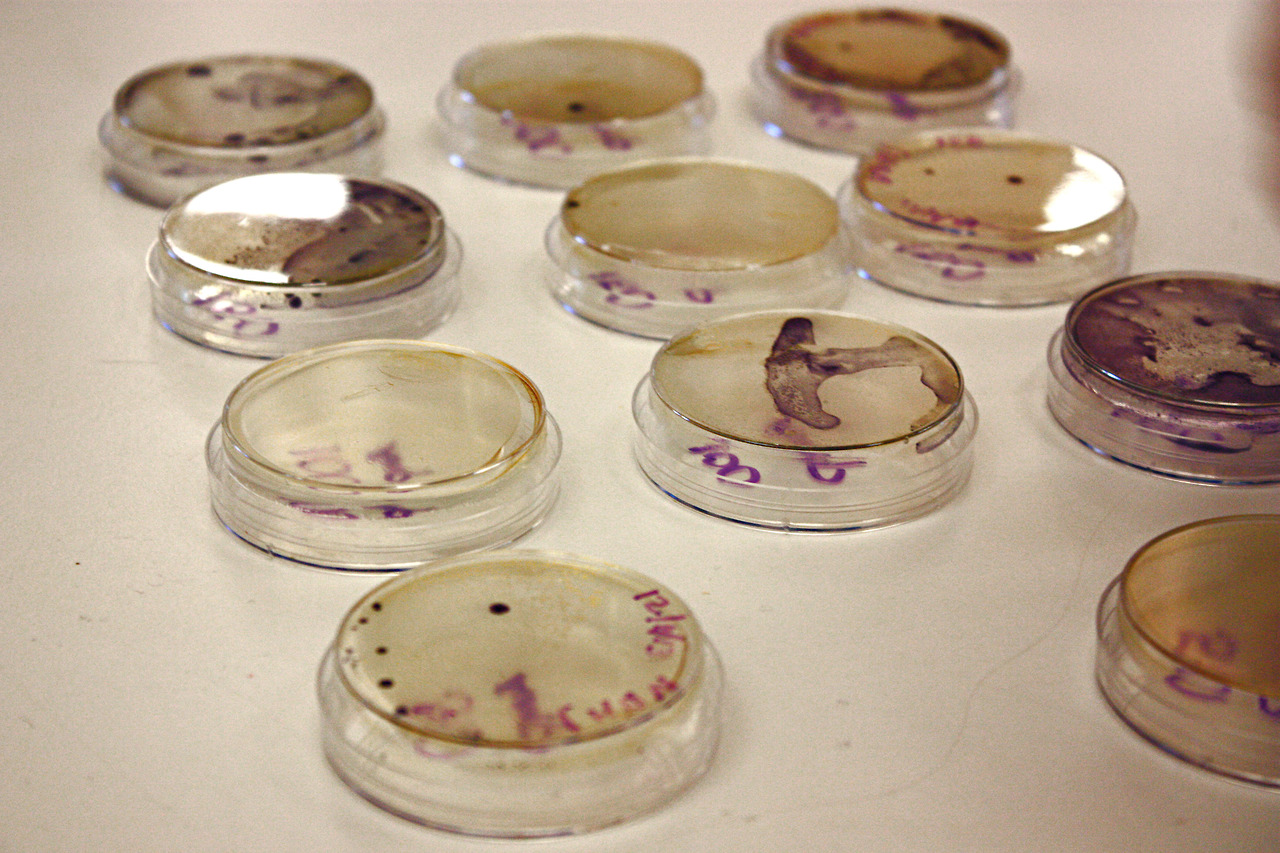

Pictured: Howard, a co-teacher and Faculty Advisor, presenting on the necessary functions of a cohesive iGEM team.
November 18

Pictured: Top: Howard draws a diagram of the parts of a biological device.
Bottom: a buffer is added to the gel after it sets.
November 20

Pictured: Top: Howard describes the inner workings of the lac operon.
November 25

Pictured: Left: Joey illustrates how the backbone of a plasmid can be cut to accept new parts.
Right: Aiden pours a gel as Liam and Mason look on.

Pictured: Top: Howard draws (and vigorously circles) a ribosome binding site.
Bottom: Howard demonstrates the process of making amp plates.
Decmber 9

Pictured: The team intently examines the working autoclave.
December 11

Pictured: Top: Nate, Aiden and Liam put their plasmids on ice.
Bottom: The plasmids are put on our ampicillin plates.
December 16

Top: Howard explains truth tables.
Bottom: Our E. coli is purple!
December 18

Top: Joey details the standardized DNA sequences that allow plasmids to be cut.
Bottom-left: Noa and Jenny work on the transformation lab.

Pictured: a comb is inserted into the not-yet-solidified gel to create wells.
January 8

Pictured: Joey and Howard divide reagents.
January 13
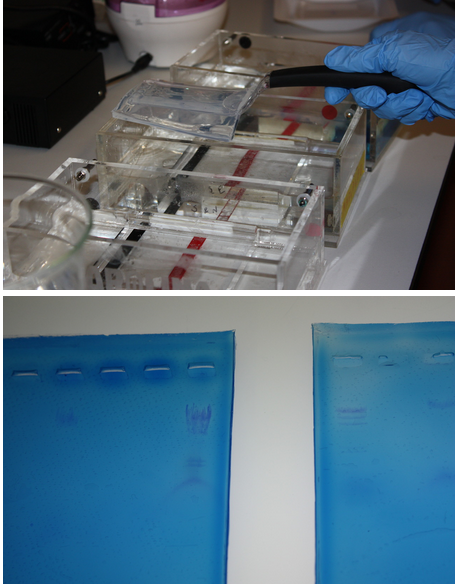
Pictured:
Top: Noa removes a gel from the electrophoresis chamber.
Bottom: Our results were highly visible when the gels were placed on a light box.
January 15

Pictured:
Nate, Aiden, and a blurry Liam fill a gel’s wells.
January 22
We dedicated the whole meeting to practicing with a restriction digest.

Pictured:
Top: Liam and Aiden present their findings.
Bottom: Noa and Jenny set up before sharing theirs.
January 27

Pictured:
Top: Joey and Howard review the process of writing a circuit.
Bottom: Lilly begins to draft a circuit on the whiteboard.
January 29

Pictured:
Top: Summer discusses her proposed circuit.
Bottom: The final list of ideas to be discussed and voted on. (Voting results coming soon…)

Pictured: Liam reviewing his proposed circuit for the project, just before the team splits into smaller groups to research various parts.
April 28
April 29
May 6
May 8
We ordered more gBlocks from IDT. We will carefully rewrite the protocol for digestion and ligation of the parts that go into the linearized pSB3A1 so transformation can be successful with a future attempt. Also will run concurrent positive control in the future to avoid our inability to analyze any future failures.
May 14
May 15
So we have the expression plasmid in NEB 10 beta cells on agar supplemented with amp. The next step is to grow up colonies form the agar plate in LB broth. This will give us a large supply of plasmids. We will miniprep the liquid culture getting bare plasmids for contranformation of DB21 DE3 competent cells with both plasmids. By using Western blot we confirm presence of RiAFP in supernatant and lysate. What did we do right this time. In the ligation step, we were very careful to use the correct proportion of DNA from the inserts (Parts A and B) and the vector (linearized pSB1A3). We used 50 ng of vector DNA and made use of a calculator on the NEB web site to calculate the amt of insert DNA to use. There should be a 3 to 1 molar excess of insert DNA to vector DNA to ensure that all the linearized vector is fully occupied by insert DNA. The calculation used the nucleotide lengths of the inserts (about 450 bp for each part) and the vector (2155 bp) to make the calculation. Also in an incubation in ligation we incorrectly used 22 degrees C instead of room temperature originally. This was corrected in the lates transformation.
May 18
The next step is to miniprep the transformed cells. Then we cotransform the two plasmids into DB21 DE3 competent cells. Our device includes an expression vector containing RiAFP cloned downstream of the T7 promoter. DB21 DE3 competent cells carry a chromosomal copy of the phage T7 RNA Polymerase gene, which is controlled by a lac promoter. When inducer (IPTG) is added, T7 RNA Polymerase is expressed and becomes dedicated to transcription of RiAFP.
Plasmid pLG575 includes the coding regions for proteins HlyB and HlyD.
May 20
May 22
Currently we have liquid cultures of NEB cells with the expression plasmid and liquid cultures of NEB cells with the secretion plasmid. At this point we are poised to do minipreps on both types of cultures.
The minipreps will give us bare plasmids that in turn will be used to transform BL21 DE3 E coli cells. These are chemically competent E. coli cells suitable for transformation and T7 promoter driven expression of RiAFP expression. The two plasmids will be used to cotransform BL21 DE3 cells. Following cotransformation, expression of RiAFP will be induced by IPTG.
May 24
There will be other analyzes that we'll also do:
• Effect of each plasmid and both plasmids on the growth rate of BL21 DE3 cells (by turbidity analysis using spectrophotmometer
• Gel electrophoresis of some of the intermediate constructs
May 24
UV transilluminator (302 nm) equipped with a camera capable of integration
To view and photograph a gel on the UV transilluminator, use a video camera, CCD (Charged Couple Device) camera, or a cooled CCD camera with ethidium bromide filter or band pass filter encompassing the emission maxima
(590 nm) of the stain.
May 27
So how 'bout we order:
InVision™ His-Tag In-Gel Staining Kit
InVision™ His-tag In-gel Stain is supplied as a 1X ready-to-use staining reagent (500 mL) [enough to run 20 mini gels]
BenchMark™ His-tagged Protein Standard (125 µL) is also included in the kit
NuPAGE® Novex® 10% Bis-Tris Gels (pre-cast polyacrylamide gels)
May 28
We have ordered from LifeTechnologies Invision in-gel stain, a latter, and precast gels for confirming the expression and secretion of the protein. These experiments will be done with Steph Hayes at Pam Silver's lab at Harvard Medical School.
Keep your fingers crossed that everything goes right and we'll be in the home stretch. Still need to do data analysis, the Wiki, our poster and the presentation so we need everybody at battle stations a while longer.
Finally, we need to do more minipreps more growing up liquid cultures, more transformations so please avail yourself of the opportunity to get your hands wet in the lab. Its fun, its instructional, and you help is really neede
May 29
• cotransformation of Bl21 De 3 cells
• miniprep of plasmids
• making agar plates
• making liquid cultures
• planning measurement of oD of culture for growth curves
May 31
If the cotransformation works our next step is to grow up the cotransformed cells in LB broth and then induce them with IPTG to express and secrete RiAFP.
Remember we plan on determining the influence on, the other and both plasmids on the growth of BL21 DE3 cells using the optical density of the liquid culture on the spectrophotometer. If time before the close of the Wiki permits we might also do digestions of the plasmids and look at their electrophoresis patterns.
June 2
We are having trouble with the cotransformation and we are worried that the plasmid DNA we are getting from the minipreps was too little. As a benchmark, it is good to have more than 50 nanograms/microliter of plasmid DNA.
We have two plasmids: pLG575 and the expression plasmid. We have 5 nanograms/microliter of this plasmid. Obviously very low. The other plasmid is the expression plasmid. We have 47 nanograms/microliter of this plasmid. Good.
We can adjust the miniprep to increase the concentration of pLG575 DNA,
The puzzle is that we should have gotten transformation of the expression plasmid. So we still have to look at the transformation procedure.
June 6
• cotransformation with the original pLG575 plasmid from Utah State and the expression plasmid
• transformation of pUC19 positive control plasmid
• transformation with pLG575 plasmid obtained by miniprep using modifications to increase concentration
• transformation of the pPURP plasmid from the BioBuilder lab exercise
All in all, maddeningly frustrating, but we will persevere.
June 7
pPURP is the plasmid from BioBuilders and pUC19 is the positive control plasmid sent with the competent cells by NEB.


We mixed new stock solution of ampicillin and chloramphenicol and carefully checked the concentrations in the LB agar, so I am really bewildered by the discrepancies between the expected and observed results.
June 10
June 11
Results from Steph’s Lab

Disregarding one or two anomalies, the analysis indicates two things:
The BL21 DE3 competent cells don't appear to be competent. Keeping them on dry ice may not have been able to maintain a stable -80 degree centigrade temp allowing them to become incapable of transformation (see column pUC19). Please note that the prior batch of competent cells from NEB (NEB 10 beta cells) were transformed by the pLG575 plasmids we recieved from Utah State and the plasmid we constructed from the gBlocks form IDT. I did not observe thawing of the BL23 DE3 cells. The plasmid DNA we obtained by miniprep were not able to transform any competent cells (see last three columns). The cause of the failure of the miniprep remains uncertain.
June 12
We have plasmids with the expression cassette (amp resistant) and the secretion cassette (chloramphenicol resistant), respectively in transformed NEB 10 beta cells. This is where we reached a stone wall. Using minipreps, we were unable to cotransform BL21 DE3 cells and induce those cells with IPTG to express and secrete RiAFP. We have an in-gel stain that will enable us to detect RiAFP in cytoplasm and culture media standing by and ready to use (takes about 3 hours to do).
Under the guidance of Steph, team members performed transformations using several E coli strains and different plasmids (the results summarized in an email message sent to all yesterday). The analysis of those results suggest that the commercial competent cells from NEB (BL21 D3) were degrade by unstable maintenance of -80 C temp and that our miniprep procedure was flawed.
It is easy to overcome the competent cell flaw. Not so easy is pin pointing what's wrong with the miniprep. Since we have amp and chloramphenicol resistant NEB 10 cells in the Bio 3 frig, we can analysis the plasmids in those cells and figure out a strategy to go forward. We have in the frig in Bio 3 agar plates and LB broth cultures of the NEB 10 beta cells which are amp resistant cells andchloramphenicol resistant cells meaning they were transformed by the expression and secretion plasmids, respectively. I will bring them to the AlbertsLab tomorrow for analysis confirming the presence of the appropriate plasmids in the cells.
Needless to say, the clock is ticking and it will be a challenge to finish all the lab work. The Wiki closes on 20 June, but the lab work can go on beyond that date and any new data can be incorporated in the Poster and the PowerPoint slide deck. Even then time may run out despite the best efforts.
June 13
Here is new revised approach:
we need a plasmid which contains these components and BioBrick standard prefix and suffix (not the RFC 23):
• T7 promoter
• RBS
• RiAFP coding sequence
• HlyA coding sequence
• 6x His marker
• double terminator
We insert (using digestion and ligation) the above fragment into a linearized plasmid with a resistance marker other than chloramphenicol (the secretion plasmid is chloramphenicol resistant). We then use Invision in gel stain to detect presence of RiAFP in cytoplasm and media.
We'll be working up to the last minute: here's the timeline:
• Receive new part from IDT: 19 June
• Insert new part into linearized plasmid: 19 June
• Transform competent cells to grow up stock of plasmid: 20 June
• Grow up liquid culture of transformant: 21 June
• Miniprep of plasmid DNA: 22 June
• Transform BL21 DE3 cells: 22-23 June
• Grow up transformed BL21 DE3 cells and induce expression and secretion of RiAFP: 23-24 June
• Use Invision system to detect RiAFP in fractions: 25 June
• Analyze results: 25-26 June
June 14
BioBrick Standard Protein Coding Region Prefix /T7 Promoter /RBS /RiAFP/HlyA /Double terminator /BioBrick Standard Protein Coding Region Suffix
The DNA sequences for the components could be found in the Registry. So it was not difficult to design the part. It was 864 base pairs in length. It is fast and simple to order a 864 base pair gBlock fragment form IDT (double-stranded, sequence-verified genomic blocks that ship in only a few working days for affordable and easy gene construction).
The first step in ordering a gBlock is automated review by screening tools and expert review by IDT scientists of entered sequences for characteristics that may interfere with synthesis. This review found that there were sequence repetitions longer than 8 base pairs that comprise a combined 91% of the sequences within a window of 70 base pairs beginning at base pair 235. This structure would interfere with synthesis. So the IDT gene product specialist optimized the sequence for expression in E coli and compatible with the gBlock synthesis process. Optimization theoretically just changes codon usage without changing the proteins coded. Importantly, the EcoRI and PstI sites on the 5’ and 3’ ends of this gene were maintained compatible with BioBrick standard 3A assembly. However, Noa at Steph's lab ran the optimized new sequence thru the seqbuilder program and identified that the optimization process put in another Pst1 restriction site at position 637 in addition to the appropriate one at position 863 (the one compatible with the BioBrick standard). Having the secondPstI restriction site breaks the BioBrick standard and makes correct assembly impossible. Following the finding of this glitch, the fragment was reoptimizedremoving the PstI site at 836. The part was order for synthesis and delivery as soon as possible.

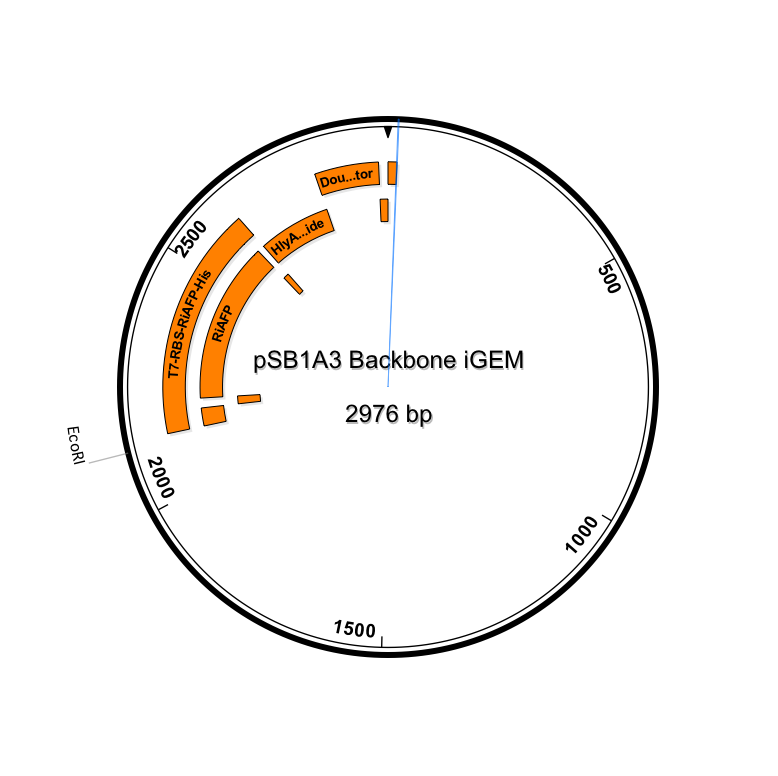
New sequence outlined in SeqBuilder (click to enlarge)
The plan is to insert the new part into pSB1A3 and again do cotransformation of BL21 DE3 cells with the two plasmids. Then we would induce thecotransformants with IPTG to express and secrete RiAFP. Finally, using theInvision In-gel Staining Kit we would analyze the media and BL21 DE3 cell lyzatefor RiAFP. This would mean we work up to the very last minute before the Jamboree but we are determined to successfully complete the project.
One last problem is a reliable source of pLG575. Because our mini-centrifuge has a max speed of 4000 rpm (as compared to the recommended of 12000 rpm) our yield of pLG575 from Qiagen miniprep is abysmal (on Nanodrop: 5 ug/uL) The pLG575 generously sent to us by our advisor at Utah State, Asif Rahman, on Nanodrop showed 260 ug/ul. I'll ask Asif to send us another tube of pLG575 from his lab. Also, we will do the miniprep with Steph using a centrifuge of appropriate speed.
So that's our plan! The team is up to the challenge. In the lull while we wait for the new part, we will work on the Wiki due 20 June. and work on the Poster and our 20 minute Presentation.